

Abstract
Introduction: Gene variants in the apolipoprotein L-1 (APOL1) gene are strong modifiers for the development of chronic kidney disease in individuals of African descent and are associated with progression of renal disease and albuminuria in cross-sectional studies of individuals with sickle cell anemia (SCA). While the association of APOL1 with albuminuria in older SCA patients is established, it is unclear whether participants with APOL1 G1 (rs73885319/ rs6090145) and G2 (rs71785313) variants (Ashley-Koch Br J Hematol 2011; Kormann Br J Haematol 2017) are more likely to develop albuminuria early in life. We hypothesized that individuals with SCA with the APOL1 G1 and G2 variants experience albuminuria at a higher rate and at a younger age than individuals without these APOL1 mutations.
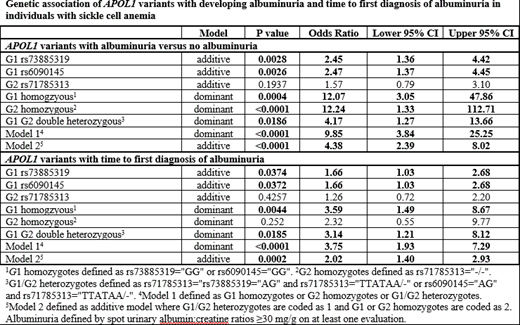
Methods: APOL1 G1 (rs73885319/ rs6090145) and G2 (rs71785313) variants were identified from whole genome sequence (WGS) data for individuals with SCA (HbSS or HbSβ0 thalassemia) enrolled in the longitudinal Sickle Cell Clinical Research and Intervention Program (SCCRIP; NCT02098863) (Hankins et al, Pediatr Blood Cancer 2018). WGS data were generated by aligning paired-end 150 bp reads to the GRCh38 human reference using the Burrows-Wheeler Aligner (BWA-ALN v0.7.12) and the GATK best-practices workflow implemented in GATK v3.4.0. We modeled the time to first albuminuria diagnosis defined by abnormal urine albumin:creatinine ratio (>30mg/g). Covariates included age, sex, and hydroxyurea therapy. We considered two risk models. Model 1 defined high risk APOL1 mutation as either APOL1 G1 homozygotes or G2 homozygotes or G1/G2 double heterozygotes. Model 2 was defined additively wherein G1 or G2 homozygotes were assumed to confer more risk compared to G1/G2 double heterozygotes.
Results: In 285 individuals with SCA, 93% (n=266) with HbSS and 7% (n=19) with HbSβ0 thalassemia, 14% (n=41) experienced albuminuria at a mean age of 11.4 (±3.3) years. In total, 11% (n=32) of this SCA cohort had an APOL1 mutation; 6% (n=17) were G1/G2 double heterozygote, 4% (n=11) were G1 homozygotes, and 1% (n=4) were G2 homozygotes. Among the 32 participants with an APOL1 mutation, 40% (n=13) had albuminuria as compared to 11% (n=28) of the 253 participants without an APOL1 mutation (OR: 9.85, 95% CI 3.84-25.25). There was also a significant association with albuminuria based on the additive genetic model (OR: 4.38, 95% CI 2.39 -8.02) (Table). In a survival analysis, participants with an APOL1 mutation had a hazard ratio (HR) of 3.75 (95% CI: 1.93-7.29, p<0.0001) associated with time to diagnosis of albuminuria compared with individuals with other non-risk APOL1 genotypes. The mean age for individuals having albuminuria with APOL1 G1/G2 risk alleles was 9.8 (±3.2) years, roughly 2 years younger than those without the risk alleles (11.8 ±4.7 years) (Satterthwaite t value=3.10, p=0.003). APOL1 G1 alleles also contributed individual risk of albuminuria (HR=1.66, p=0.0374).
Summary: Our analyses found that pediatric individuals with SCA who have APOL1 two risk alleles are at high risk of developing albuminuria which many will experience early in childhood. This important finding has significant implications for clinical care. First, using a genetic screening program for all patients with SCAC can identify individuals with APOL1 mutations. Second, individuals with SCA and an APOL1 mutation should be screened for albuminuria during childhood. Finally, early initiation of therapeutic strategies to ameliorate renal dysfunction needs to be evaluated in the presence of APOL1 mutations.
Hankins:Global Blood Therapeutics: Research Funding; Novartis: Research Funding; NCQA: Consultancy; bluebird bio: Consultancy. Estepp:ASH Scholar: Research Funding; Daiichi Sankyo: Consultancy; NHLBI: Research Funding; Global Blood Therapeutics: Consultancy, Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal